
Let op: De volgende informatie voor distributeurs is opgesteld op basis van de Duitse- en de Nederlandse markt en geldt uitsluitend binnen de EU. Indien van toepassing zijn er aanvullende, landspecifieke voorschriften die in acht moeten worden genomen, die hier niet kunnen worden behandeld.
De overgangsperiode van de nieuwe Medical Devices Regulation 2017/745 (MDR), de Europese Regeling Medische Hulpmiddelen, loopt op 26 mei 2021 af. Naast veel nieuwe eisen en verplichtingen voor fabrikanten van medische hulpmiddelen, bevat dit ook bepaalde verplichtingen waar distributeurs van medische hulpmiddelen aan moet voldoen. Om u te informeren hebben we op deze pagina de belangrijkste informatie over de omgang met Ofa producten verzameld. Deze informatie is specifiek gericht op Ofa producten, die behoren tot klasse I en niet steriel zijn. Bij het omgaan met producten van andere fabrikanten kunnen er andere verplichtingen zijn. Daarom vindt u de volledige tekst van de MDR en twee richtlijnen van de DGIV hiernaast gelinkt.
Belangrijk: Voor alle producten die u vóór 26 mei 2021 heeft ontvangen, geldt een rechtsgeldige houdbaarheidstermijn tot en met 26.05.2025, mits de houdbaarheid van onze producten dit toelaat. Voor deze producten zijn ook de afgegeven conformiteitsverklaringen volgens de Medical Device Directive (93/42/EEG), de Richtlijn Medische Hulpmiddelen, nog steeds geldig. Voor alle producten die na 26 mei 2021 zijn ontvangen gelden de nieuwe regels. Voor deze producten stellen wij een EU-conformiteitsverklaring op conform de Medical Devices Regulation 2017/745 (MDR).
De taken van een distributeur: Zorgplicht - Inspectieplicht - Informeerplicht
Deze informatie is gebaseerd op artikel 14 van de MDR. In samenwerking met de fabrikant zijn deze distributeursverplichtingen bedoeld om ervoor te zorgen dat alleen wettelijk conforme en veilige medische hulpmiddelen aan de eindgebruikers worden geleverd.
De verplichtingen van een distributeur zijn onder te verdelen in drie soorten. In de zorgplicht staat dat de gespecialiseerde distributeur vanaf 26 mei 2021 alle geldende regels van de MDR met de nodige zorgvuldigheid in acht moet nemen.
De inspectie- en informatieplicht omvat elk verschillende taken, die we hieronder nader zullen toelichten. Sommige moeten worden ingevuld voordat ze aan de patiënt worden verstrekt, andere hebben betrekking op de tijd daarna.
Links and Downloads
Informatiebladen en checklist:
MDR-informatieblad voor Ofa-distributeurs
Checklist inspectieverplichtingen
Conformiteitsverklaringen:
Conformiteitsverklaringen Steunkousen
Conformiteitsverklaringen Rondbrei compressie
Conformiteitsverklaringen Vlakbrei compressie
Conformiteitsverklaringen Orthopedie
Conformiteitsverklaringen Accessoires
Gebruiksaanwijzingen:
Gebruiksaanwijzingen Steun- en Compressiekousen
Gebruiksaanwijzingen Bandages en Orthesen
De MDR om te lezen:
DGIHV richtlijnen voor implementatie:
MDR-gids van de DGIHV voor fabrikanten van op maat gemaakte producten (Duits)
1. Voorafgaande aan uitgifte aan de patiënt: Conformiteit controleren
Voordat een Ofa-product aan de patiënt afgeleverd wordt, moeten een aantal zaken gecontroleerd worden. Deze controles dienen zorgvuldig gedocumenteerd te worden. Niet elk product hoeft te worden gecontroleerd, maar er dient een steekproefprocedure te worden gehanteerd, die representatief is voor de producten die aangeboden worden. Idealiter vinden deze controles direct op de ontvangstafdeling plaats, voordat de producten - voorafgaande aan de uiteindelijke afgifte aan de patiënt - opgeslagen worden.
Voordat de fabrikant of distributeur een Ofa-product aan een van haar klanten verstrekt, dient gecontroleerd te worden dat betreffende product voldoet aan de nieuwe MDR-richtlijn. Dit doen ze door ervoor te zorgen dat er een conformiteitsverklaring voor het product is afgegeven in overeenstemming met de MDR. Voor klasse I-producten is hiervoor geen aangemelde instantie vereist, maar de fabrikant Ofa Bamberg geeft onder eigen verantwoordelijkheid een conformiteitsverklaring af. Met de conformiteitsverklaring verklaren de fabrikant en de distributeur dat de medische hulpmiddelen die eronder vallen voldoen aan alle eisen van de voor deze hulpmiddelen geldende voorschriften van de Regeling Medische Hulpmiddelen 2017/745 (MDR). De ondertekende conformiteitsverklaring is tevens de basis voor de CE-markering; zonder conformiteitsverklaring mag dit niet worden uitgevoerd.
U vindt de Ofa conformiteitsverklaringen in het downloadoverzicht aan de rechterkant.

Naast de CE-markering dient de distributeur enkele andere markeringen en informatie op onze Ofa producten te controleren. Deze vindt men op de verpakking, het verpakkingsetiket of in de gebruiksaanwijzing. Wat gecontroleerd moet worden:
Is het product (verpakking/verpakkingsetiket) voorzien van de volgende markeringen?
1. Naam of handelsnaam van het product
2. Informatie waaruit de gebruiker kan opmaken waar het product over gaat
3. De naam van de fabrikant en het adres van zijn maatschappelijke zetel
4. Een indicatie dat het product een medisch hulpmiddel is
5. Informatie over opslag en hantering
6. Vervaldatum (productiedatum in het geval van op maat gemaakte producten)
De distributeur is verplicht zich te houden aan de opslag- en verwerkingsinstructies zoals vermeld op de product etikettering. Er moet passend bewijs kunnen worden geleverd van naleving van de opslag- en hanteringsvoorwaarden.

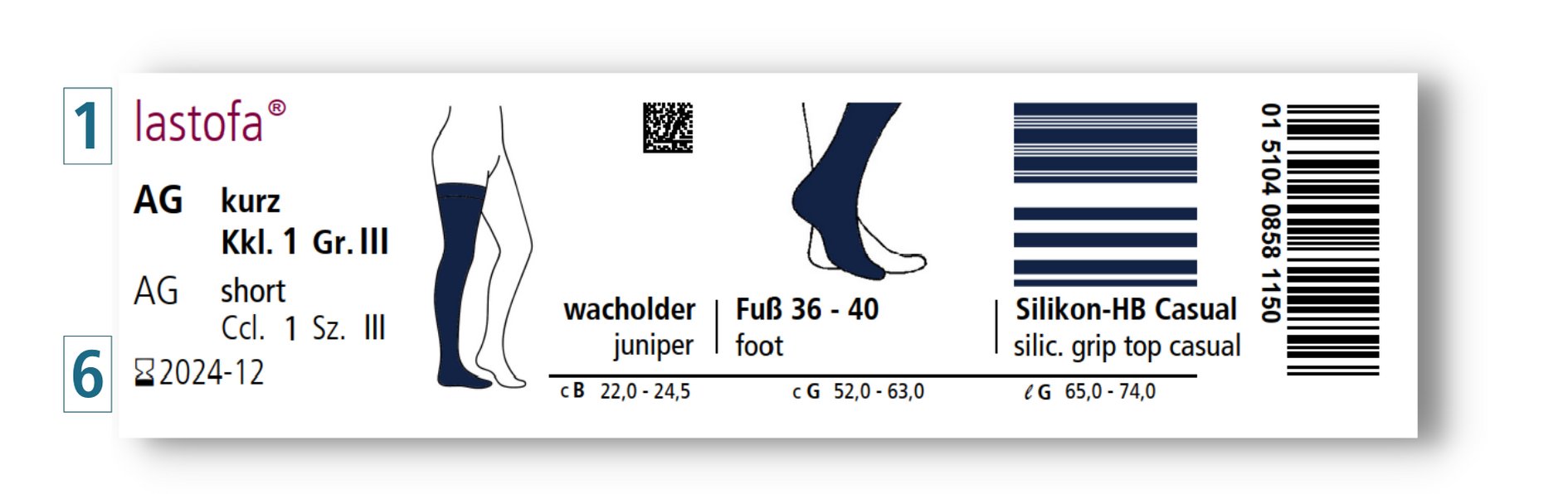
Op onze verpakkingen vindt u ook het volgende nieuwe etiket:

Zo ziet u in één oogopslag dat dit product bedoeld is voor eenmalige afgifte aan één patiënt en door precies deze patiënt meerdere keren mag worden gebruikt, conform de gebruiksaanwijzing.
Zitten er gebruiksaanwijzingen bij?
Alle Ofa-producten worden met gedetailleerde gebruiksaanwijzingen geleverd. Deze kunnen ook montagehandleidingen voor de vakhandel bevatten (meestal op de binnenzijde van de omslag). Met de gebruiksaanwijzing voldoen wij aan onze verplichting als fabrikant van medische producten om op begrijpelijke wijze uitleg te geven over het gebruik van onze producten en de mogelijke risico's tijdens het gebruik. Dit houdt in dat in het geval van distributie in het buitenland, deze informatie beschikbaar is in de respectievelijke officiële taal van het land.
Ook stellen we een digitale versie van onze gebruiksaanwijzingen online beschikbaar voor patiënten. De links naar de gebruiksaanwijzing uit de verschillende productgebieden vindt u in het overzicht rechtsboven.
Bevatten de gebruiksaanwijzingen alle informatie die de MDR vereist?
Dit bevat:
1. Naam van het product
2. Naam van de fabrikant en het adres
3. Datum van uitgave of herziening van de gebruiksaanwijzing
4. Informatie over opslag en hantering
5. Het beoogde gebruik van het product
6. Informatie over gebruik, zorg, indicaties, contra-indicaties, bijwerkingen, restrisico's
7. Informatie over de verplichting om de fabrikant en de bevoegde autoriteiten bij ernstige incidenten te informeren

Om de traceerbaarheid van producten te vereenvoudigen, is de zogenaamde Unique Device Identification (UDI) geïntroduceerd. Dit bestaat onder meer uit een barcode of datamatrixcode die bepaalde informatie codeert. Deze informatie omvat het GTIN (Global Trade Identification Number, (01)), dat het product op modelniveau uniek identificeert (bijv. Dynamics Carpaal Orthese, maat 1, rechts), evenals het LOT-nummer (10) en de vervaldatum (17) van het betreffende individuele product. Daarnaast moet deze informatie dan ook nog eens goed leesbaar naast de code worden vermeld. De datamatrixcode en de informatie in gewoon schrift vormen samen de volledige UDI.
Voor klasse I-producten is deze markering niet verplicht tot 26 mei 2025. Bij Ofa Bamberg zullen we dit jaar echter al de UDI-markering toepassen. De uitvoering staat gepland tot het einde van het jaar. Om u een idee te geven van hoe deze wijziging eruit zal zien, vindt u hier een voorbeeldlabel.
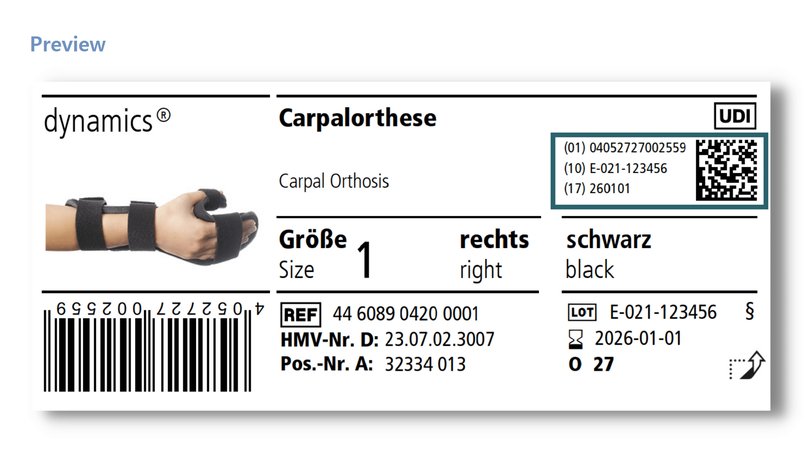
Op basis van de gegevens van de UDI is nadere informatie over de producten te vinden in de centrale Europese databank "EUDAMED". De volledige functionaliteit van deze database zal echter waarschijnlijk pas in mei 2022 beschikbaar zijn.
Indien de distrubuteur na controle van de CE-markering, conformiteitsverklaring en gebruiksaanwijzing tot de conclusie komt dat deze niet voldoen aan de eisen van de MDR, mag die distrubuteur het product niet aan patiënten vrijgeven. Bovendien is hij/zij in dit geval verplicht Ofa Bamberg als fabrikant onmiddellijk op de hoogte te stellen via vp@ofa.de.
Als het product een ernstig risico inhoudt (zie onderstaande definitie), dient de distributeur ook de bevoegde hogere federale overheid te informeren. In Nederland is dit de Inspectie Gezondheidszorg en Jeugd (IGJ). U vindt de meldingsformulieren ook online op de IGJ-website. Meer gedetailleerde informatie over wat een incident, een ernstig incident of een ernstige bedreiging voor de volksgezondheid is, vindt u in het volgende tabblad.
Bovendien moet u de niet-conformiteit van het product documenteren. Meer hierover in het tabblad "Klachtenregister".
De eerdere zorgplichten, zoals het niet geven van beschadigde of defecte producten aan patiënten, blijven gelden.
2. Na levering aan de patiënt: follow-up en monitoring
Conformiteit is gecontroleerd, het product is afgeleverd. Dit is echter niet het einde van de taken van de distributeur. Als eerste aanspreekpunt voor patiënten is de distributeur een belangrijk onderdeel van de informatie- en meldketen bij klachten, technische of applicatieproblemen en incidenten. Maar wat is een incident precies?
In de loop van de MDR is de definitie van incident gewijzigd. Daarnaast zijn de termen ernstig incident en ernstige bedreiging voor de volksgezondheid geïntroduceerd. Deze termen worden gedefinieerd in artikel 2 (64.-66.) van de MDR:
Incident
betekent elke storing of verslechtering van de kenmerken of prestaties van een hulpmiddel dat op de markt wordt aangeboden, met inbegrip van gebruiksfouten als gevolg van ergonomische kenmerken, evenals elke ontoereikendheid in de door de fabrikant verstrekte informatie en elk ongewenst neveneffect.
Ernstig incident
betekent elk incident dat direct of indirect leidde, had kunnen leiden of zou kunnen leiden tot een van de volgende zaken:
a) het overlijden van een patiënt, gebruiker of andere persoon,
b) de tijdelijke of blijvende ernstige verslechtering van de gezondheidstoestand van een patiënt, gebruiker of ander persoon,
c) een ernstige bedreiging voor de volksgezondheid.
Ernstige bedreiging voor de volksgezondheid
betekent een gebeurtenis die kan leiden tot een dreigend risico op overlijden, een ernstige verslechtering van de gezondheidstoestand van een persoon of een ernstige ziekte, die onmiddellijke herstelmaatregelen kan vereisen en die aanzienlijke morbiditeit of mortaliteit bij de mens kan veroorzaken, of die ongebruikelijk of onverwacht is voor de opgegeven plaats en tijd.
De distributeur heeft de plicht om met de fabrikant en met de autoriteiten samen te werken om gevaren af te wenden en corrigerende maatregelen mogelijk te maken. Als de distributeur van mening is of redenen heeft om aan te nemen dat een product een ernstig risico inhoudt, dient hij/zij de fabrikant (vp@ofa.de) en de bevoegde autoriteiten van de lidstaten waar hij/zij het product op de markt heeft gebracht, onmiddellijk op de hoogte te stellen. Daarbij dienen in het bijzonder details over de niet-naleving en over eventuele corrigerende maatregelen die zijn genomen, gegeven te worden.
De distributeur van medische hulpmiddelen is, op grond van de MDR, verplicht om bepaalde zaken te documenteren en dit bewijsmateriaal op verzoek ter beschikking te stellen aan de fabrikant of de bevoegde autoriteiten. Naast de bovengenoemde documentatie van steekproeven moet de distributeur van medische hulpmiddelen een klachtenregister bijhouden.
Dus als de distributeur van medische hulpmiddelen klachten ontvangt van beroepsbeoefenaren in de gezondheidszorg, patiënten of gebruikers over vermoedelijke incidenten met betrekking tot een product dat hij/zij levert, de distributeur van medische hulpmiddelen deze niet alleen onmiddellijk door testuren naar Ofa Bamberg als fabrikant (vp@ofa.de) en, indien van toepassing, naar de bevoegde autoriteiten , maar leg hij/zij deze klachten ook vast in een register. Naast klachten moeten ook niet-conforme producten, terugroepingen en terugnames in dit register worden vastgelegd. Het klachtenregister moet regelmatig naar Ofa Bamberg als fabrikant gestuurd worden.
Daarbij dient de documentatie gedetailleerd en begrijpelijk voor een derde partij (bijvoorbeeld een autoriteit) te zijn. De autoriteiten kunnen ook monsters van de producten bij de distributeur opvragen, die hij/zij dan gratis dient te verstrekken.
Om de traceerbaarheid van producten te garanderen, moet u de volgende informatie minimaal 10 jaar aan de autoriteiten kunnen verstrekken:
- alle marktdeelnemers (fabrikanten, distributeurs, importeurs) van wie u een product rechtstreeks hebt verkregen
- alle marktdeelnemers (andere distributeurs, geen patiënten!) aan wie u rechtstreeks een product heeft geleverd
- alle zorginstellingen of zorgprofessionals aan wie u rechtstreeks een product heeft geleverd.
Bovendien bent u als distributeur verplicht om met ons als fabrikant en met de bevoegde autoriteiten samen te werken als corrigerende maatregelen of productterugroepingen nodig zijn.
Wellicht ook interessant voor u:
Reclame voor medische producten
De distributeur mag alleen afbeeldingen en informatie in advertenties en informatie over producten gebruiken, die door de fabrikant voor het betreffende product zijn bedoeld. Afwijkingen zoals oude of andere versies, die op enigerlei wijze zouden kunnen leiden tot het wekken van een verkeerde indruk met betrekking tot het product en zijn eigenschappen, zijn verboden.
De distributeur verstrekt geen informatie in mondelinge of schriftelijke vorm aan patiënten die als "misleidende informatie" kan worden beschouwd.
Dit omvat, maar is niet beperkt tot, informatie welke:
- functies en eigenschappen toekennen aan het product die het niet bezit
- een verkeerde indruk geven met betrekking tot de behandeling of diagnose en de functies of eigenschappen van het product
- de gebruiker of patiënt niet informeren over de verwachte risico's die gepaard gaan met het gebruik van het apparaat in overeenstemming met het beoogde doel
- ander gebruik van het hulpmiddel voorstellen dan vermeld als onderdeel van het beoogde doel waarvoor de conformiteitsbeoordeling is uitgevoerd
Actuele afbeeldingen van onze producten kunt u bij ons aanvragen; tel.: 0485-385123 of via email: info@ofa-nederland.nl.
MDR - Dit verandert er voor consumenten
Sinds 26 mei mei 2021 is de Medical Device Regulation (kortweg MDR) van toepassing op fabrikanten van medische hulpmiddelen zoals Ofa Bamberg. De Europa brede verordening regelt onder meer het veilig op de markt brengen van medische hulpmiddelen voor menselijk gebruik. Welke veranderingen de MDR voor de consument met zich meebrengt, hebben we voor u op een rijtje gezet.
De MDR regelt de volledige levenscyclus van een medisch hulpmiddel, van creatie tot verwijdering. Het doel is om de bescherming van de gebruiker te waarborgen. Deze bescherming moet nu alle economische actoren en zorginstellingen in heel Europa omvatten.
Nieuwe labels
Daarom kregen alle medische hulpmiddelen bij de inwerkingtreding van de MDR op 26 mei 2021 een keurmerk dat bevestigt dat het inderdaad om een medisch hulpmiddel gaat. Dit "MD"-teken is dan ook een indicatie van het testen volgens de nieuwe EU-eisen. Het merkteken moet op het product zichtbaar zijn - in het geval van Ofa Bamberg-producten staat bijvoorbeeld het MD-symbool op de etiketten.
Daarnaast moet op de producten of verpakkingen duidelijk zichtbaar zijn wie de fabrikant van het product is. Zo wordt duidelijk wie verantwoordelijk is voor het product en wie aansprakelijk is bij twijfel. Hiervoor helpt een klein, gekleurd fabriekssymbool bij de oriëntatie.
Bovendien zijn de gebruiksaanwijzingen nog gedetailleerder, b.v. ze bevatten nu informatie over de juiste verwijdering van medische hulpmiddelen.
Houdbaarheid op het eerste gezicht
Een andere eis van de MDR is dat elk product een houdbaarheidsdatum heeft, die te herkennen is aan een zandlopersymbool. Het product kan tot deze datum veilig worden gebruikt. Bij Ofa Bamberg zijn bijvoorbeeld steunkousen en compressiekousen tot drie jaar houdbaar, orthesen tot vijf jaar na fabricagedatum. Als er geen mededeling wordt gedaan over de houdbaarheidsdatum, moet de fabricagedatum, de productiedatum van het product worden vermeld. Deze is gemarkeerd met de lege fabriek.
Meldingsplicht incidenten
Vanwege wettelijke voorschriften binnen de EU zijn patiënten en gebruikers verplicht elk ernstig incident tijdens het gebruik van een medisch hulpmiddel onmiddellijk te melden aan zowel de fabrikant als de bevoegde nationale autoriteit. In Nederland is dit de Inspectie Gezondheidszorg en Jeugd (IGJ).